Флуоресцентно-микроскопические методы оценки функционального состояния и выживаемости нейронов
Регистрация внутриклеточной концентрации свободных ионов Са, Nа и Н
Измерения концентрации свободных ионов Са2+ ([Ca2]i)

а также их спектры возбуждения флуоресценции (сдвинутые влево кривые на графиках) и спектры испускания флуоресценции (сдвинутые вправо кривые на графиках). Для Fura-2 показана химическая структура собственно индикаторной формы, тогда как для Fluo-3 и Rhod показаны структуры их ацетоксиметиловых эфиров. Из представленных индикаторов только Fura-2 является двухволновым, поэтому на графике спектры Fura-2 представлены
Изменения концентрации ионов Са2+ в клетке отражаются на динамике, по-видимому, всех внутриклеточных процессов, включая биохимические реакции, структурные перестройки, функционирование и подвижность органелл, регуляцию генома, а также межклеточные взаимодействия12345678. С нарушений кальциевого гомеостаза зачастую начинаются нарушения функционирования всей клетки. Пути предотвращения нарушений Са2+-гомеостаза и поиск способов его нормализации после того, как дизрегуляция произошла, являются одним из лидирующих направлений предупреждения гибели клеток91011. После того как было установлено, что митохондрии не только в изолированном состоянии, но и внутри клетки способны поглощать огромные (в масштабах клетки) количества Са2+, интерес к регуляторной и патогенной роли Са2+ в клетках возрос еще больше1213. Это особенно актуально для нейронов, поскольку данный тип клеток содержит в плазматической мембране и синаптических окончаниях большое число разнообразных потенциал- и лигандуправляемых Са2+ каналов14. При нарушениях в центральной нервной системе происходит избыточная стимуляция глутаматных рецепторов, влекущая за собой двумя парами кривых: А – спектры Са2+-связанной формы; В – спектры в безкальциевом буфере. Для индикаторов семейства Rhod показаны также заместители (R5, R6) и места их локализации, позволяющие изменять константу диссоциации комплексов с Са2+ без изменения их флуоресцентных спектров (см. также цветную вклейку) массивный вход Са2+ в цитоплазму и последующую гибель клеток1516. В настоящее время общепризнана решающая роль митохондрий в способности нейронов поддерживать кальциевый гомеостаз и восстанавливать его после патологических воздействий глутамата171819202122. Потеря митохондриями способности поглощать избыток Са2+ из цитоплазмы, обеспечивать аденозинтрифосфорной кислотой (АТФ) Na+, K+– и Са2+-насосы плазматической мембраны и ретикулума, сохранять целостность внешней мембраны, удерживая цитохром С и факторы апоптоза от попадания в цитоплазму, имеет катастрофические последствия для клетки.
Первые надежные измерения изменений концентрации Са2+ в клетках были выполнены с применением Са2+-чувствительного белка акварина23. Однако настоящий прорыв в этой области наступил после того, как были синтезированы индикаторы, в которых хелаторы двух- и трехвалентных катионов, структурно аналогичные EDTA (ЭДТА – этилендиаминтетрауксусная кислота) и EGTA, соединены с флуоресцирующими молекулами таким образом, что связывание Са2+ значительно изменяет оптические свойства флуорофора2425. В настоящее время на рынке доступно уже несколько десятков таких синтетических индикаторов, отличающихся главным образом аффинностью по отношению к Са2+, спектральными свойствами флуорофора и в некоторых случаях способностью накапливаться во внутриклеточных компартментах (преимущественно в митохондриях). Свойства Са2+-индикаторов, примеры применения, включая список литературы и галерею иллюстраций, можно найти на сайте фирмы Invitrogen (http://probes.invitrogen.com/handbook/). Скрупулезный анализ свойств основных классов флуоресцентных Са2+-индикаторов дан в обзоре26.
Структурные формулы и спектры некоторых наиболее часто используемых синтетических Са2+-индикаторов показаны на рис. 1.
Для облегчения проникновения индикаторов внутрь клетки карбоксильные группы, которые несут отрицательные заряды и поэтому препятствуют прохождению молекулы сквозь фосфолипидный бислой клеточной мембраны, предварительно защищают, превращая их в ацетоксиметиловые эфиры. Ацетоксиметильная форма не является индикатором, а становится таковым внутри клетки, где эфирные связи гидролизуются эстеразами. На рис. 1 структура Fura-2 представлена в качестве пентааниона, т.е. в Са2+-чувствительной индикаторной форме.
Структуры Fluo-3 и индикаторов семейства Rhod изображены в форме ацетоксиметиловых эфиров. На примере Rhod показано также, каким образом, модифицируя молекулу индикатора, можно получать его аналоги, обладающие разной Са2+-связывающей способностью (см. вставку таблицы на рис. 1). Например, Rhod-2 имеет константу диссоциации Kd=0,6 мкМ и поэтому пригоден для измерения [Са2+]i в диапазоне 0,06-6 мкМ. Для измерения более высоких концентраций необходимо брать аналоги с более высокими Kd, к примеру, Rhod-FF или Rhod-5N (см. рис. 1), которые пригодны для измерения [Са2+]i в диапазоне 2-200 мкМ и выше 30 мкМ соответственно.
Методики загрузки клеток ацетоксиметиловыми эфирами Са2+индикаторов обычно представлены на сайтах фирм-производителей. Как правило, рекомендуют подбирать условия в зависимости от вида клеточной культуры, ее возраста, температуры инкубации, типа индикатора. Так, Rhod-2 (и его аналоги) имеет положительный заряд на ароматическом кольце, в результате чего его АМ-форма способна накапливаться внутри митохондрий27. Это может осложнить интерпретацию данных, если Rhod-2 использовать как индикатор для всей клетки, но может быть и преимуществом, когда требуется определить концентрацию Са2+ именно в митохондриях. Необходимо подчеркнуть, что при измерениях концентрации Са2+ с помощью синтетических индикаторов избирательно в каком-либо внутриклеточном компартменте требуется очень тщательный подбор условий загрузки индикатора в клетку2829.
В тех случаях, когда нужно измерить среднюю по клетке концентрацию Са2+, типичная методика, в том числе и для нейронов, заключается в инкубации клеток с АМ-формами индикаторов в солевом буфере в течение 40-60 мин при комнатной температуре. Обычно АМ-формы хранят в виде 1-2 мМ растворов в ДМСО и при загрузке клеток добавляют их в буфер в концентрациях 1-5 мкМ. Для предотвращения образования мицелл АМ-формы во внеклеточном буфере и облегчения проникновения сквозь клеточную мембрану рекомендуют проводить загрузку в присутствии «мягкого» неионного детергента Pluronic F-127 (0,02%). Загрузку проводят в том же буфере, который впоследствии используют для измерений. Иногда, если того требуют условия эксперимента, клетки загружают в СО2-инкубаторе, добавляя АМ-форму соответствующего индикатора в инкубационную среду. Состав солевого буфера при измерениях внутриклеточной концентрации Са2+ и других ионов может несколько различаться в разных лабораториях, но все буферы представляют собой вариант раствора Рингера. Солевой состав буфера, используемого в нашей лаборатории, таков (мМ): 130 NaCl, 5,4 KCl, 20 HEPES, 5 Glucose; 2 Ca, 1 Mg, рН=7,4.
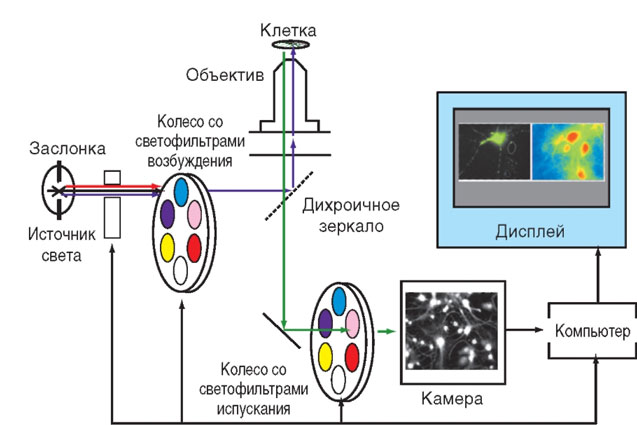
на основе инвертированного микроскопа, системы автоматической смены колес со светофильтрами в каналах возбуждения и испускания флуоресценции, камеры на основе матрицы с переносом заряда (CCD-камеры) и компьютера с программами управления колесами и камерой, а также программами получения, сохранения и анализа изображений (рисунок в измененном виде позаимствован из Chiesa et al., 2001).
Принципиальная схема флуоресцентно-микроскопической установки для мониторинга сигналов индивидуальных клеток показана на рис. 2.
Источником света служит газоразрядная дуговая лампа (ксеноновая или ртутная), имеющая спектр излучения в УФ/видимой области, достаточно широкий для возбуждения всех известных флуорофоров. Заслонка между лампой и колесом со светофильтрами перекрывает излучение на то время, когда не происходит регистрация сигнала, предотвращая фоторазрушение исследуемого объекта. Фильтровое колесо, перемещая светофильтры в соответствии с заданной программой, вырезает из всего спектра излучения лампы свет тех длин волн, которые необходимы для возбуждения используемого флуоресцентного индикатора. Дихроичное зеркало (часто его еще называют дихроичным кубиком из-за формы оправы, в которую оно вставлено) отражает возбуждающий свет в сторону объектива и пропускает излучение образца (окрашенной клетки) в сторону детектора. Объектив фокусирует возбуждающий свет на образце и собирает его флуоресценцию, направляя ее сквозь дихроичное зеркало на детектор. На показанной схеме флуоресценция, прежде чем достигнуть детектора, пропускается через светофильтр, встроенный в эмиссионное фильтровое колесо, которое управляется синхронно с колесом возбуждения. В более дешевых установках используют стационарный, сменяемый вручную, светофильтр. Детекторами служат скоростные высокочувствительные камеры, имеющие в качестве световоспринимающего устройства фотодиодные матрицы. Сигнал матрицы преобразуется компьютером в изображение для мониторинга в реальном режиме времени и хранения информации для последующей обработки. Самой сложной и дорогой частью флуоресцентно-микроскопической установки является микроскоп, представленный на схеме в виде двух деталей: дихроичного зеркала и объектива. Источник света и устройство смены фильтров, камера, компьютер с программным обеспечением обычно представляют собой модули, пригодные в комплектации со многими микроскопами, либо поставляются как единая система основными фирмами-производителями микроскопов (см., например, сайты Carl Zeiss, Nicon, Olympus). Термин «внутриклеточная концентрации свободных ионов кальция ([Са2+]i)» вошел в научный обиход тогда, когда измерения были возможны почти исключительно на клеточных популяциях, поскольку спектральные свойства флуоресцирующей репортерной группы (молярный коэффициент экстинкции, квантовый выход флуоресценции), а также чувствительность детекторов были недостаточными для регистрации сигналов индивидуальных клеток и тем более внутриклеточных компартментов. Измерения выполняли в основном на суспензиях клеток с помощью спектроили фильтровых фотофлуориметров. Естественно, в этом случае сигнал был усреднен по всей клеточной популяции, и величина [Са2+]i трактовалась как одинаковая во всем объеме клетки.
Синтез Fura-2 и Indo-130, а затем Fluo-3 и Rhod-231, имеющих гораздо больший молярный коэффициент экстинкции и динамический диапазон (кратность изменения сигнала индикатора в Са2+-связанной форме по отношению к свободной), а также доступность высокочувствительных камер для регистрации изображения, сделал возможным измерения [Са2+]i в индивидуальных клетках и даже их компартментах. В 1990-е годы появились работы, в которых было показано, что концентрация свободного Са2+ в сарко- и эндоплазматическом ретикулуме может на порядки превосходить таковую в среднем по клетке323334. Изобретение флуоресцентномикроскопических установок, с высокой чувствительностью регистрирующих излучение камер и конфокальных микроскопов, позволило показать, что в ответ на возбуждение клетки изменения [Са2+] внутри клетки могут распространяться волнами35 или проявляться в виде локальных скачков, вспышек (sparks) 3637. Изменения концентрации Са2+ в митохондриях (как по амплитуде, так и кинетически) также существенно отличаются от таковых в среднем по клетке3839.
Арсенал средств, применяемых при изучении внутриклеточной Са2+-сигнализации, принципиально изменился, когда на основе генетически модифицированных флуоресцентных белков из медуз и других морских организмов и Са2+-связывающего белка кальмодулина были сконструированы сенсоры внутриклеточного Са2+40. Генетические конструкты флуоресцентных белков были получены и для отслеживания других важнейших параметров клеток и мониторинга поведения конкретных белков4142. Главное преимущество флуоресцентных индикаторных белков по сравнению с синтетическими Са2+-индикаторами – возможность создания конструктов, включающих адресный пептид, обеспечивающий доставку химеры в интересующий внутриклеточный компартмент. Благодаря таким конструктам были показаны пространственное переплетение и функциональная кооперация эндоплазматического ретикулума и митохондрий434445.
В нейронах [Са2+]i чаще всего измеряют там, где регистрируется максимальный сигнал индикатора – в теле нейрона. Измерение [Са2+] в аксонах, дендритах и тем более синапсах требует более чувствительной и дорогой техники конфокальной микроскопии.
Из вышесказанного следует, что «внутриклеточная концентрация свободных ионов Са2+ ([Са2+]i)» – достаточно условный термин, под которым скрывается параметр, усредненный по всем внутриклеточным компартментам. Часто, когда измерения ограничены сомой нейрона, данную проблему стараются обойти тоже терминологически, называя этот параметр «концентрацией свободных ионов Са2+ в цитозоле ([Са2+]c)» (см., например,46). Следует, однако, иметь в виду, что во многих типах клеток, а в нейронах в особенности, значительную часть клетки занимает ядро. Таким образом, сигнал индикатора, собираемый со всего объема клетки (точнее – со всей фокальной поверхности), представляет собой сигнал, усредненный между цито- и нуклеоплазмой. В ядерной оболочке имеются поры, размер которых позволяет свободно диффундировать из цитозоля в ядро и обратно молекулам размером до 20-40 кДа47. Этого более чем достаточно для быстрого прохождения любых неорганических ионов и синтетических индикаторов и выравнивания их концентраций в цитозоле и ядре. Однако сопоставление физико-химических и спектральных свойств синтетических Са2+-индикаторов в ядре и цитозоле показало, что свойства индикаторов в этих компартментах не всегда одинаковы484950. Это обстоятельство, к сожалению, трудно поддается учету на практике. Таким образом, замена термина «внутриклеточная концентрация Са2+» на термин «концентрация Са2+ в цитозоле» лишь скрывает проблему, не решая ее. Мы в дальнейшем будем придерживаться термина, которому присвоили обозначение [Са2+]i.
Описанная проблема может быть решена путем применения генно-инженерных Са2+-сенсоров, так как их локализация определяется наличием адресной последовательности. Отсутствие таковой означает локализацию сенсора там, где он синтезируется, – в цитозоле или в цитозоле и ядре, если размеры сенсора не препятствуют прохождению сквозь ядерную пору. Несмотря на такое преимущество, по сравнению с синтетическими Са2+-индикаторами генетические конструкты пока не вытеснили такие индикаторы, как Fura-2, Fluo-3 и их аналоги по двум основным причинам, приведенным ниже.
- Трансфекция клеток соответствующими векторами и экспрессия в этих клетках сенсоров – хуже воспроизводимая и гораздо более длительная (обычно 2-3-дневная) процедура, чем загрузка клеток синтетическими индикаторами (см. выше).
- Сенсоры Са2+ на основе флуоресцентных белков подвержены значительной рН-зависимости сигнала и применимы только тогда, когда рН в том компартменте, где они локализованы, изменяется несущественно.
Оба эти ограничения, к сожалению, особенно актуальны в нейронах. Во-первых, будучи дифференцированными клетками, они с трудом поддаются трансфекции. Во-вторых, стимуляция рецепторов, сопряженных с ионными каналами, особенно гиперстимуляция ионотропных глутаматных рецепторов при патологических процессах, вызывает не только мощный вход Са2+, но и сильное закисление цитоплазмы и митохондрий51.
Флуоресцентные Са2+-индикаторы по зависимости их спектров от концентрации Са2+ подразделяют на одно- и двухволновые. Это свойство присуще как синтетическим индикаторам, так и генноинженерным сенсорам. Заключается оно в том, что у одноволновых индикаторов в ответ на увеличение [Са2+] растет только интенсивность флуоресценции без изменения максимума спектров возбуждения и испускания. В качестве примера таких индикаторов на рис. 1 приведены Fluo-3 и Rhod-2. У двухволновых индикаторов в ответ на увеличение [Са2+] наряду с изменением интенсивности происходит сдвиг спектра возбуждения (спектры Fura-2 на рис.1) или эмиссии (Indo-1). Благодаря сдвигу спектров находят такие длины волн, интенсивность которых изменяется в противоположных направлениях. Для Fura-2 флуоресценция по мере образования комплекса с Са2+ растет в области длин волн 330-360 нм в спектре возбуждения и падает в области длин волн 360-400 нм (см. рис. 1). Эти изменения флуоресценции отражают изменения соотношения концентраций свободной и связанной с Са2+ формы индикатора. Двухволновые индикаторы часто называют рэйшиометрическими (ratiometric), поскольку результаты измерения их сигналов удобно представлять как отношение интенсивностей при таких длинах волн, при которых интенсивности (F) изменяются в противоположных направлениях. Для Fura-2 это F340 и F380 (нм) соответственно для Са2+-связанной и свободной формы индикатора (см. рис. 5.1). Уравнения, связывающие интенсивности с [Са2+]i, и описание процедуры калибровки можно найти во многих статьях и монографиях. Синтез Fura-2 и Indo-1 впервые описан и проанализирован наряду с преимуществами рэйшиометрических индикаторов в работе52.
Для одноволнового индикатора или для любой из длин волн двухволнового индикатора уравнение выглядит следующим образом:
[Ca2+]i = Kd х (F – Fmin) / (Fmax – F) (1)где Kd – константа диссоциации комплекса индикатора с Са2+ (мкМ), F – текущее значение интенсивности флуоресценции (отн. ед.); Fmin и Fmax – интенсивности флуоресценции индикатора соответственно в среде без кальция и в Са2+-связанной форме.
Значения Fmin определяют, добавляя к клеткам Са2+-ионофор (чаще всего иономицин) в присутствии высокой концентрации хелатора Са2+ EGTA (5-10 мМ). Иономицин селективно пермибилизует все клеточные мембраны для Са2+, сохраняя их целостность, а EGTA связывает Са2+, вытягивая его из клетки и «отбирая» у индикатора, превращая индикатор в бескальциевую форму.
Значения Fmax определяют, насыщая индикатор кальцием путем замещения раствора с EGTA таким же, но содержащим вместо EGTA высокую концентрацию Са2+ (2-5 мМ). Совокупность процедур определения Fmin и Fmax называют калибровкой и выполняют в конце эксперимента либо в отдельном эксперименте в идентичных условиях.
Для двухволнового индикатора (на примере Fura-2) процедура калибровки аналогична, но уравнение выглядит немного иначе:
[Ca2+]i = Kd х (R – Rmin) / (Rmax – R) x (Fmin / Fmax)380 (2)где R – текущее отношение F340/F380; Rmin и Rmax – значения R, определяемые во время калибровки совершенно так же, как Fmin и Fmax в случае одноволнового индикатора. Отношение (Fmin/Fmax)380 соответствует значениям интенсивности при возбуждении флуоресценции на 380 нм, получаемым при калибровке в бескальциевом буфере (Fmin)380 и в присутствии насыщающей концентрации Са2+ (Fmax)380. Необходимо подчеркнуть, что подстрочные индексы «min» и «max» относятся к концентрациям Са2+, а не к величинам (Fmin)380 или (Fmax)380. В присутствии EGTA (Fmin)380 больше, чем (Fmax)380 (см. рис. 1).
![Изменения [Ca2+]i](https://cmi.to/wp-content/uploads/2020/04/izmeneniya_Ca1.png)
Изменения [Ca2+] представлены как относительные изменения рэйшиометрического сигнала Fura-2 (F340/F380) и относительные изменения сигнала Fluo3-FF (F490/F0), где F490 - текущее значение сигнала, а F0 - значение в покоящихся клетках, предшествовавшее добавлению глутамата (Glu, 100 мкМ). Значения Kd взяты на сайте Invitrogen
Выше было отмечено, что выбор индикатора для регистрации изменений [Са2+]i зависит от того, каковы ожидаемые изменения [Са2+]i. Так, при воздействии на нейроны токсических доз глутамата (Glu) наблюдается двухфазный характер изменений [Са2+]i (рис. 3).
Рис. 3 четко показывает, что высокоаффинный индикатор Fura-2 (Kd=0,22 мкМ) дает значительное увеличение сигнала (F340/F380) в момент добавления Glu, но относительно небольшой рост во время второй фазы подъема (отмечено стрелкой). Низкоаффинный индикатор Fluo-3FF (Kd=10 мкМ) демонстрирует иное соотношение сигналов: небольшой ответ на добавление Glu, но зато значительный рост во время второй фазы, названной отсроченной кальциевой дизрегуляцией5354. Различие сигналов обусловлено следующим. В момент добавления Glu [Са2+] возрастает примерно до 1-1,5 мкМ, что соответствует почти всему диапазону [Са2+] который можно измерить с помощью Fura-2 (не более 2,5-3,0 мкМ). Именно поэтому дальнейший рост [Са2+]i происходящий во время отсроченной кальциевой дизрегуляции, вызывает лишь небольшое увеличение сигнала. Низкоаффинный Fluo-3FF, в соответствии с величиной его комплекса с Са2+ (Kd=10 мкМ), пригоден для измерения [Са2+]i в диапазоне от 1 до 100 мкМ. В результате этого первоначальный рост его сигнала (F490/F0) выглядит весьма слабым, зато величина вторичного подъема F490/F0 свидетельствует о том, что во время отсроченной кальциевой дизрегуляции [Са2+]i возрастает до десятков мкМ, что, безусловно, является патологическим событием для клетки5556. Приведенный пример демонстрирует, что индикатор необходимо подбирать таким образом, чтобы Kd соответствовала примерно середине ожидаемого диапазона изменений [Са2+]i.
Флуоресцентно-микроскопическая система и многие методические приемы, разработанные и описанные выше для изучения Са2+-гомеостаза клеток, можно успешно применять и для исследования внутриклеточного гомеостаза других катионов. Ниже в качестве таковых рассмотрены примеры измерения концентраций ионов N+ и H+.
Изменения концентрации свободных ионов Na и H+ (рН)
Измерение внутриклеточного рН (рН)
Почему мы измеряем внутриклеточный pHi?
Активность ферментов. Известно, что активность большинства ферментов зависит от pHi. Так, показано, что гликолиз ускоряется при повышении pHi, активность фосфофруктокиназы изменяется пропорционально сдвигам в pHi 57 .
Ионизация субстратов. Ионизация определенных аминокислотных участков субстратов ферментов, которая происходит при определенном внутриклеточном рН, оказывает влияние на активность ферментов. Например, многие ферменты используют ионизированные субстраты АТФ (MgATP2-, MgADP-, ADP2-, ADP3-). При рН=7,0 ионизация этих субстратов составляет 28-36%, поэтому уменьшение рН на 0,5 ЕД приводит к падению их концентраций более чем в 2 раза58.
Метаболизм кислот и оснований. рН участвует в механизмах, контролирующих метаболизм клеток или тканей. Эти процессы являются взаимосвязанными, поскольку известно, что ионы Н+ сами по себе участвуют во многих реакциях, например:
Lactate- + NAD+ ↔ pyruvate + NADP-H + H+, (3)
где NAD – никотинамидадениндинуклеотид; NADP – никотинамидадениндинуклеотидфосфат, NADP-H – восстановленная форма NADP.
В качестве показательного примера такой взаимозависимости можно привести следующее: удаление лактата печенью отражает удаление протонов, образованных в результате мышечного гликолиза (цикл Кори).
Движение метаболитов через мембраны. Определение рН снаружи и внутри клеточной мембраны вместе с мембранным потенциалом может указывать на направление транспорта ионов или метаболитов.
Методы измерения рН
Существует несколько методов измерения рН Они представлены ниже.
- Использование слабокислых или щелочных индикаторов. Этот метод основан на том, что для некоторых слабых кислот или оснований проницаемость биологических мембран для неионизированных форм во много раз больше, чем для ионизированных, поэтому концентрация неионизированных форм должна быть эквивалентна в двух компартментах (вне- и внутриклеточных), в то время как количество ионизированных форм зависит от рН в каждом компартменте. Слабые кислоты должны появляться в высокой концентрации в более щелочном компартменте, слабые основания, наоборот, – в более кислом. При использовании данного метода необходимо учитывать и другие факторы: не должно быть связывания и активного транспорта используемых соединений; константа диссоциации (Kd) должна быть одинаковой в изучаемых компартментах; индикаторы не должны быть токсичными и подвергаться метаболизму; необходимо быть уверенным в том, что распределение индикаторов в компартментах достигло во время измерения равновесия. С учетом всего сказанного этот метод в настоящее время не применяют.
- Метод ядерной магнитно-резонансной спектроскопии. Этот метод более привлекателен для исследователя по сравнению с предыдущим. Его принцип описан в ряде статей и основан на анализе резонансных частот ядерных спинов ионизированных групп; частоты зависят от степени ионизации, которая, в свою очередь, зависит от рН Для выполнения применяют или фосфор-31, или 2,3-фосфоглицерат, которые в конечном счете определяют метаболизм неорганического фосфата, а последний тесно связан с рН. Однако необходимо быть уверенным в том, что при изучении рН нет сигналов от наружного неорганического фосфора, т.е. рН достаточно отличается от рН0. Это характерно для сердца, печени и почек. Используют при анализе также и время продолжительности ядерного спина. Недостатком метода является необходимость применения дорогостоящего оборудования.
рН-Чувствительные красители
Существуют особые вещества, которые меняют свой цвет в зависимости от величины рН. Например, нейтральный красный при рН=6,0 – красного цвета, а при увеличении рН до 8,0 меняет свой цвет на желтый. Пики абсорбции составляют 530 и 445 нм соответственно. Этот краситель обычно используют для топографического гистохимического измерения рН в мозгу или при сокращении мышц. Основное достоинство этого метода – то, что регистрацию рН можно проводить в течение всего эксперимента: краситель реагирует очень быстро на любые изменения рН. Однако имеются и определенные недостатки: связывание с белками, влияние экстрацеллюлярной жидкости и др.
Флуоресцентные зонды

(из книги «Руководство по флуоресцентным зондам»)
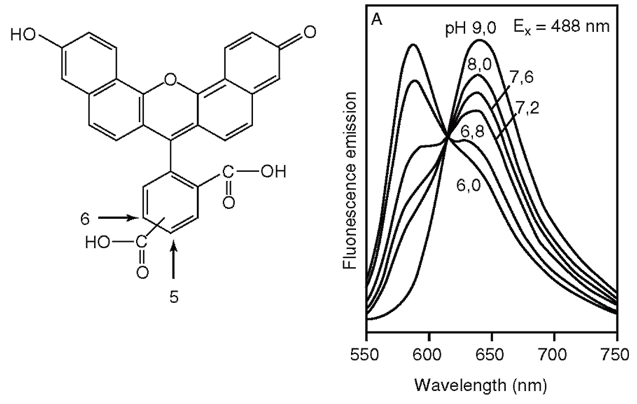
Цитозольный рН в покоящихся клетках колеблется около величины 7,0 в пределах десятых долей, поэтому использование внутриклеточных индикаторов требует выбора таких, рК которых близко к 7,0. В настоящее время наиболее популярным флуоресцентным зондом для измерения рН является BCECF (2,7-bis(carboxyethyl)-5 carboxyfluorescein) 59.
BCECF – производное флуоресцеина с тремя дополнительными карбоксилатными группами, добавленными для увеличения гидрофильности (рис. 4); к двум из них прикреплены короткие алкильные цепочки, которые позволили повысить рКа до 7,0 (для флуоресцеина рКа=6,4). BCECF, как и все флуоресцеины, является сильным флуорофором с пиками возбуждения и эмиссии на 503 и 525 нм соответственно.
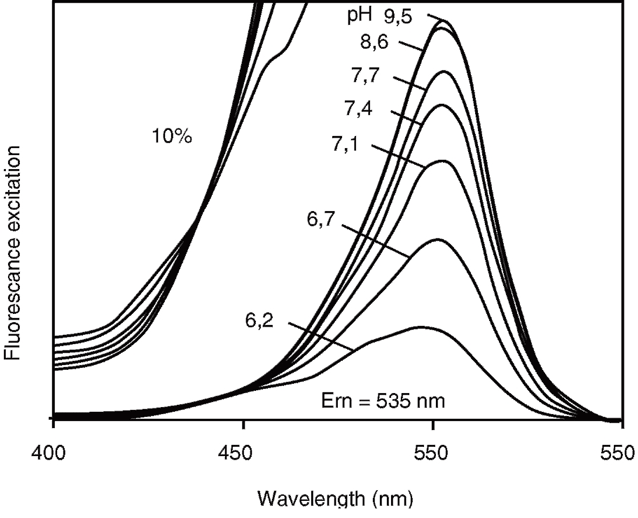
На рисунке убедительно представлено, что изобестической точкой флуоресценции зонда является диапазон 435-439 нм
Флуоресценция зонда рН-зависима (по амплитуде пиков): падает при закислении среды и увеличивается при ее защелачивании. Достоинством этого зонда является то, что при его возбуждении светом в 430-439 нм возникающая флуоресценция рН-независима (рис. 5)60. Наличие рН-независимой и рН-чувствительной части в спектре возбуждения очень важно для исследователей, поскольку отношение рН-чувствительной части к рН-нечувствительной не зависит от концентрации флуоресцентного зонда, длины оптического пути, освещенности иллюминатора, чувствительности детектора. Все эти преимущества позволили успешно применять систему изображения при работе на флуоресцентном микроскопе с единичными культивируемыми клетками61. Хотя BCECF можно вводить в клетки микроинъекцией, наибольшее распространение получил метод инкубации клеток с несколькими мкМ ацетоксиметилового эфира BCECF (BCECF/AM). Этот эфир нейтрален и гидрофобен, а поэтому мембранопроницаем и легко входит в клетку. В клетке BCECF/AM с помощью цитозольных эстераз постепенно превращается в BCECF, который является гидрофильным относительно непроникающим анионом.
Измерение рН проводили при помощи флуоресцентного зонда BCECF
Флуоресценция BCECF в клетке легко калибруется in situ с помощью нигерицина и буферных растворов с известным рН. Нигерицин – это К+, Н+-ионофор. Принцип калибровки следующий: в растворе с нигерицином (5 мкМ) концентрация внеклеточного К+ равна концентрации внутриклеточного К+, при этом рН; равно рН0. Исследователь с помощью буферных растворов с разными величинами рН может откалибровать флуоресценцию зонда.
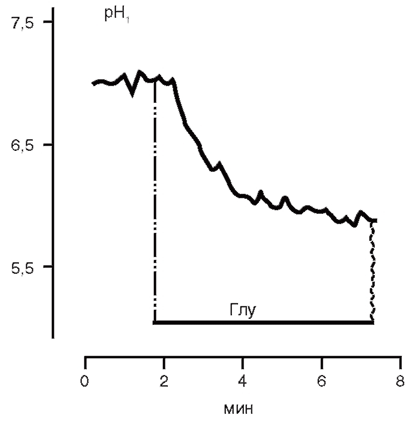
В качестве примера приводим результаты собственных измерений рН;, проведенных на культивируемых зернистых нейронах мозжечка при кратковременном воздействии глутамата. Предварительно был приготовлен маточный раствор BCECF/AM в ДМСО (1 мМ). Этот раствор можно хранить в морозильнике при -20 °С и использовать многократно. В день исследования в буферный раствор с клетками вносили 2-4 мкл исходного раствора, клетки инкубировали 30-40 мин при 37 °С, затем 3 раза отмывали буферным раствором, не содержащим флуоресцентный зонд. На столик инвертированного микроскопа устанавливали покровное стекло, и на клетки подавали попеременно возбуждающий флуоресценцию свет 440 и 490 нм, флуоресценцию регистрировали при 535 нм. После окончания эксперимента проводили калибровку рН сигнала (рис. 6). На рисунке видно, что глутамат (100 мкМ) вызывает закисление цитозоля нейронов.
На рисунке видны pH-чувствительные и рН-независимые спектры зонда
Наряду с BCECF в исследованиях используют также SNARF-1. Этот рН-индикатор является нафтофлуоресцином (рис. 7).
В отличие от BCECF, SNARF-1 – двухволновой по флуоресценции (588 и 635 нм) и одноволновой при возбуждении (514 нм). рКа зонда составляет 7,5. Этот зонд широко используют при проведении исследований с помощью проточной цитометрии или конфокального микроскопа. Методы загрузки зонда и его калибровки такие же, как и для BCECF.

Приведены все данные, полученные с помощью YFP-конструкций, как относительные изменения флуоресценции
В настоящее время установлено, что сигнальные системы во внутриклеточных органеллах, например в цитозоле и митохондриях, во многом зависят от распределения и транспорта водородных ионов через мембрану органелл. Однако измерение рН в этих компартментах стало возможно проводить совсем недавно после создания генно-инженерных конструкций, связанных с зеленым флуоресци рующим белком (green fluorescent protein – GFP) из Aequora victoriya. Так, в настоящее время выделены несколько рН-чувствительных белков, селективно экспрессирующих в цитозоле и митохондриях cytYFP (рНц) и mtYEP (рНм)62. В исследованиях, проведенных на культивируемых корковых нейронах, мы измерили изменения рН цитозоля и митохондрий63. Для измерения рНmir, и рНс нейроны трансфицировали с помощью реагента Lipofectamine 2000 плазмидами, кодирующими изоформы желтого флуоресцирующего белка (yellow fluorescent protein – YFP), селективно экспрессирующегося в митохондриях и цитозоле. Чувствительность mtYFP и cytYEP к изменениям рН показана на рис. 8.
![Взаимоотношения между [Ca2+]c и цитозольным и митохондриальным рН во время глутаматного воздействия](https://cmi.to/wp-content/uploads/2020/04/vsaimootnoshenie_Ca.png)
Культивируемые корковые нейроны были трансфицированы mtYFP и cytYEP и загружены флуоресцентным зондом Fura-2FF для измерения [Ca2+]c и одновременной регистрации рНц и рНм. Панели а и б представляют типичные изменения [Ca2+]c и рН, вызванные глутаматом
Первичное повышение [Са2+]c сопровождалось закислением цитозоля с последующим его защелачиванием во время развития 2-й фазы повышения [Са2+]c (см. рис.9). В то же время при первичном повышении [Са2+]c в митохондриях наблюдали транзиторное защелачивание, которое во время развития 2-й фазы увеличения первичного повышения [Са2+]c сменялось длительным закислением. Таким образом, использование указанных выше флуоресцентных зондов открывает большие возможности для изучения клеточного метаболизма и различных сигнальных систем.
Измерение внутриклеточного натрия
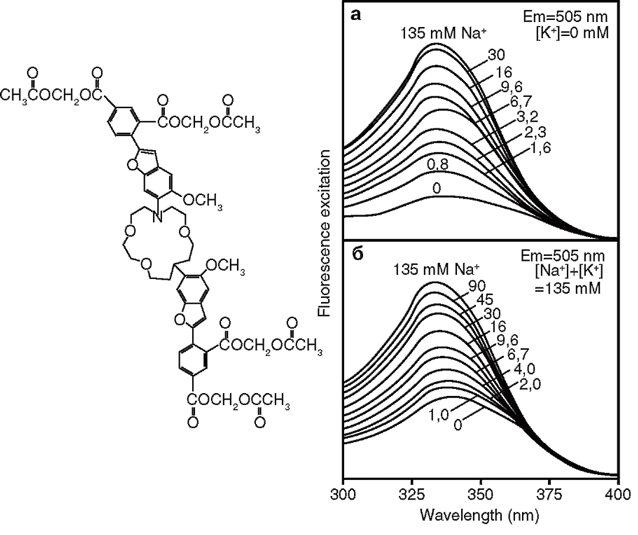
Известно, что концентрация натрия внутри клетки ([Na+]i) в 20-30 раз меньше, чем вне клетки. Именно поэтому транспорт натрия в клетку идет по электрохимическому градиенту, связанному с потенциалом плазматической мембраны, тогда как транспорт натрия из клетки идет с большой затратой энергии. Изменения [Na+]i играют важную роль в изменениях метаболизма клетки при увеличении ее активности или повреждении. Для измерения [Na+]i в настоящее время широко применяют флуоресцентные зонды, а именно – SBF]. Химическая структура зонда представлена на рис. 10.
В молекуле SBFI присоединенный к основной структуре зонда флуорофор подобен Са2+-индикатору Fura, поэтому при определении [Na+]i используют те же длины волн, что при определении [Са2+]i, а именно: при эмиссии – 510 нм, при возбуждении – 340 и 380 нм. Калибровку флуоресцентных сигналов проводят с помощью порообразующего антибиотика грамицидина, который выравнивает концентрацию Na+ по обе стороны плазматической мембраны. В качестве примера приводим собственные данные об изменениях [Na+]i в нейронах с помощью SBFI (рис. 11)
![Изменение внутриклеточной концентрации натрия ([Na+]i)](https://cmi.to/wp-content/uploads/2020/04/izmeneniya_Ca2.png)
в культивируемых зернистых клетках мозжечка крыс в ответ на разные дозы нейромедиатора глутамата (5 и 100 мкМ). Измерение [Na+]i проводили с помощью флуоресцентного красителя SBFI (Сторожевых Т.П. и др., 2007)
Оценка выживаемости (гибели) нейронов
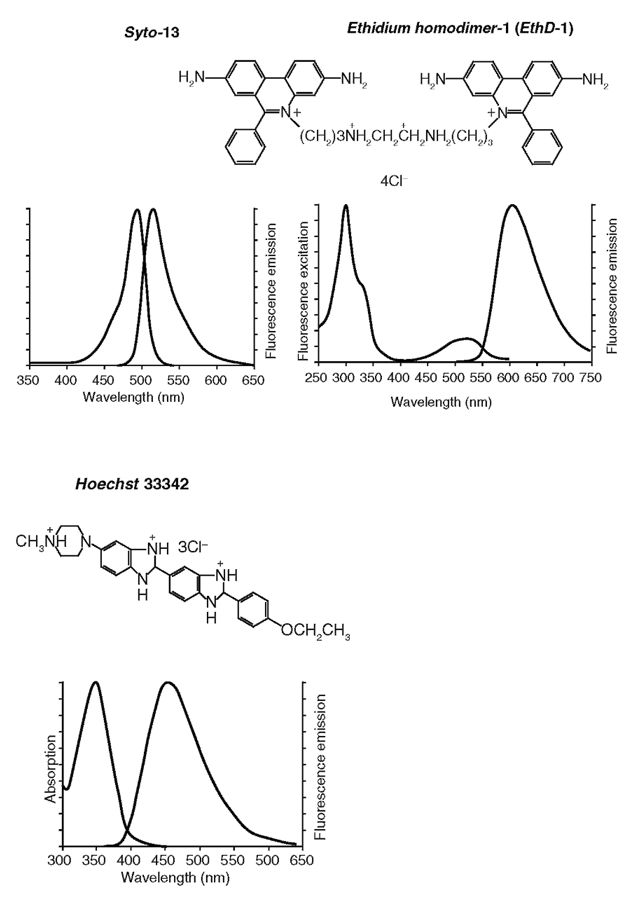
(сдвинутые вправо кривые на графиках) красителей, окрашивающих живые и умершие клетки (Hoechst 33342); только живые (Syto-13) и только мертвые (ethidium homodimer-1, EthD-1) клетки, утратившие целостность плазматической мембраны (графики и структурные формулы воспроизведены с сайта компании Invitrogen
При определении биологической активности веществ необходимым этапом считают исследование их влияния на выживаемость клеток в культуре. Доля живых и мертвых клеток в культуре или в свежевыделенных образцах (например, крови) может быть определена самыми разными способами. Методы, основанные на измерении оптических свойств самих клеток и/или проникших в них (или связанных с поверхностью) сильно поглощающих свет или флуоресцирующих зондов, обладают рядом серьезных преимуществ. В отличие от использования радиоактивных меток, работа с оптическими зондами не требует особых мер предосторожности. Оптическое оборудование и реагенты существенно дешевле (за исключением, может быть, конфокальных микроскопов), чем счетчики радиоактивности и радиоактивные препараты. Морфологические исследования флуоресцентно окрашенных клеток позволяют определить такие промежуточные стадии гибели индивидуальных клеток, которые недоступны другим методам (за исключением электронной микроскопии). Ниже мы остановимся на некоторых наиболее популярных флуоресцентных агентах, используемых при исследовании гибели нейронов в результате глутаматной нейротоксичности и при поиске нейропротекторных фармакологических средств. Кроме того, будут рассмотрены наиболее наглядные, на наш взгляд, примеры на других типах клеток, применимые и при исследованиях нейрональных культур.
Морфологические методы оценки выживаемости нейронов
Применение флуоресцентных витальных красителей
Арсенал оптических реагентов и зондов для определения выживаемости клеток, предлагаемый различными фирмами, чрезвычайно широк [см., к примеру, Invitrogen (http://probes.invitrogen.com/ handbook/)]. На рис. 12 показаны структуры, а также спектры возбуждения и испускания флуоресценции трех витальных красителей. В данном примере общим свойством всех трех красок является их способность связываться с нуклеиновыми кислотами клетки. Связывание приводит к удержанию зондов внутри клетки и многократному увеличению интенсивности их флуоресценции.
В наших экспериментах по исследованию выживаемости гранулярных нейронов мозжечка крысы, подвергнутых действию нейротоксических доз глутамата, были использованы следующие концентрации флуоресцентных зондов: Hoechst 33342 – 10 мкг/мл; Syto-13 – 1 мкМ; EthD-1 – 3 мкМ. Окрашивание проводили путем добавления маточных растворов (все зонды были растворены в ДМСО) к клеткам в солевом буфере и инкубировали в течение 25-30 мин при комнатной температуре. Затем культуры ополаскивали тем же буфером и помещали на столик микроскопа для подсчета клеток. Состав солевого буфера тот же, что и при измерениях внутриклеточной концентрации Са2+ и Na+ (см. выше).
Благодаря тому, что спектры возбуждения и эмиссии каждого из указанных выше витальных красителей сдвинуты друг относительно друга на десятки нанометров, можно возбуждать и регистрировать флуоресценцию каждого по отдельности и наблюдать индивидуальные сигналы этих зондов при их совместном применении. Различия в окрашивании разными флуоресцирующими агентами позволяют обнаружить такие состояния клетки, которые очень трудно различить между собой при применении каждого из красителей по отдельности. Особенно это показательно для идентификации промежуточных стадий гибели клетки, в частности, апоптоза. Согласно предложению Номенклатурного комитета по клеточной гибели, одним из трех необходимых и достаточных морфологических крите риев смерти клетки является потеря целостности клеточной мембраны, тогда как при апоптозе целостность мембраны сохраняется, но значительно меняется структура ядра64. Примеры живых, мертвых и апоптотических клеток показаны на рис. 13 и 14 для культуры гранулярных нейронов мозжечка крысы и мононуклеарных клеток периферической крови.
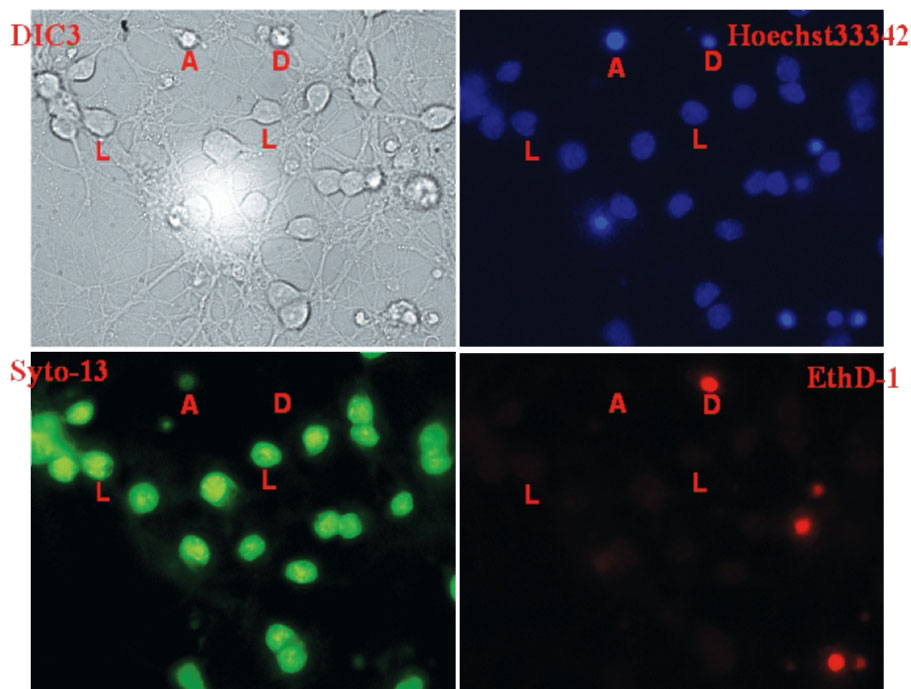
полученное на просвет в режиме дифференциального интерференционного контраста (DIC), и флуоресцентные изображения того же поля, полученные с применением флуоресцентных зондов Hoechst 33342, Syto-13 Green и этидиум гомодимер (EthD-1). Буквами помечены несколько живых нейронов (обозначены «L») и нейронов, находящихся в разных стадиях гибели (обозначены «А» и «D»). Живые (L) клетки отчетливо видны как зерна вытянутой формы с отростками (DIC), имеющие четко окрашенное ядро (Hoechst 33342), ядро и цитоплазму (Syto-13) и не имеющие окрашивания этидиум гомодимером (EthD-1). Нейроны в стадии апоптоза (А) имеют ярко светящееся сконденсированное ядро (Hoechst 33342) и отсутствуют на изображениях, полученных при регистрации флуоресценции Syto-13 и EthD-1. Мертвые (D) нейроны, в которых уже нарушена целостность плазматической мембраны, имеют небольшое пикнотическое ядро, слабо светящееся или совсем незаметное при окрашивании Hoechst 33342, но ярко светящееся при регистрации флуоресценции EthD-1; ядра всегда отсутствуют на флуоресцентном изображении клеток, окрашенных Syto-13. Представленные изображения получены с использованием масляно-иммерсионного объектива 63х/NА 1,3, трехполосного дихроичного зеркала TFT 440-500-570 и полосовых светофильтров возбуждения/эмиссии (нм): Hoechst 33342 - 360/460, Syto-13 - 488/520, EthD-1 - 550/620
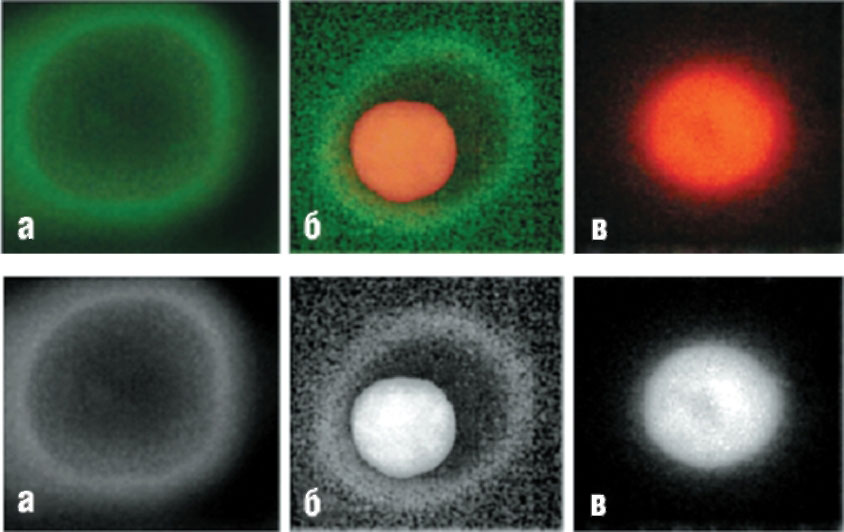
окрашенных FITCаннексином V (AnnV) и пропидий иодидом (PI). Изображения показывают три клетки, находящиеся на ранней стадии апоптоза (а), поздней стадии апоптоза (б) и в стадии некроза (в). Слабое зеленое кольцеобразное свечение соответствует AnnV, связанному с клеточной мембраной; ярко-красное свечение PI отмечает положение ядра (рисунок взят из работы Baskic et al., 2006)
Hoechst 33342 и Syto-13 способны за несколько минут проникать сквозь клеточные мембраны и окрашивать те компартменты клетки, в которых находятся нуклеиновые кислоты. Однако Hoechst 33342 связывается премущественно с ДНК, тогда как Syto-13 способен примерно одинаково связываться как с ДНК, так и с РНК. В результате этого флуоресценция Hoechst 33342 (синяя на рис. 13) более яркая в ядерной зоне, а Syto-13 (зеленая на рис. 13) примерно одинакова по всей клетке. Красное свечение этидиум гомодимера (EthD-1) наблюдается в тех клетках, в которых отсутствует Syto-13. Известно, что EthD-1 окрашивает только такие клетки, в которых нарушена целостность плазматической мембраны, что является первичным морфологическим критерием ее смерти65. Хорошо видно, что свечение Hoechst 33342 в этих клетках ограничено небольшой областью пикнотического ядра (см. рис. 13).
Буквой «А» на рис. 13 отмечена одна из клеток, находящаяся, вероятно, в стадии апоптоза. В этой клетке интенсивное свечение Hoechst 33342 отмечается только в области ядра, значительно отличается от флуоресценции в живых нейронах и пока не окрашено EthD-1. Последнее указывает на целостность клеточной мембраны. Сохранение целостности плазматической мембраны при изменении морфологии как ядра, так и клетки в целом (см. DIC-изображение (дифференционный интерференционный контраст) на рис. 13) служит признаком апоптоза66. Любопытно, что в отмеченной клетке свечение Syto-13 очень слабое и выглядит небольшим пятном, совпадающим с изображением Hoechst 33342. Возможно, эта стадия апоптоза сопряжена со значительным разрушением РНК. При нейротоксических воздействиях высоких доз глутамата наблюдают дозозависимое падение числа живых клеток и увеличение доли мертвых и находящихся в стадии апоптоза6768.
Другой пример того, как сочетание разных зондов позволяет получить уникальную информацию о стадиях гибели клетки, приведен на рис. 14, взятом из работы69. Показаны три мононуклеарных клетки периферической крови, находящиеся на разной стадии гибели.
В клетке (а) уже начались апоптотические процессы, приведшие к нарушению распределения фосфатидилсерина в фосфолипидном бислое мембраны. В результате этого значительная доля фосфатидилсерина появилась на внешней поверхности клеточной мембраны, что позволило флуоресцентно-меченному аннексину V связаться с ней, образуя кольцеобразное зеленое свечение (см. рис. 14, а). Отсутствие окрашивания ядра пропидий иодидом (PI) указывает на целостность клеточной мембраны (см. рис. 14, а), что соответствует, как было отмечено выше, критериям нахождения клетки в состоянии апоптоза70. На рис. 14, в представлена клетка, в которой ярко светится ядро, окрашенное PI, что считают одним из первичных критериев некроза. Любопытный пример представляет собой клетка на рис. 14, в. В ней отмечается одновременно окрашивание и плазматической мембраны, и ядра. Такую ситуацию трактуют как позднюю стадию апоптоза, при которой клеточная мембрана еще окончательно не распалась, но уже потеряла барьерные свойства для PI71.
Путем окрашивания культуры кортикальных нейронов акридиновым оранжевым и этидиум бромидом с последующим анализом конфокальных флуоресцентных изображений было показано, что в течение первых 2-4 ч действия глутамата превалирует некроз. Удлинение времени действия глутамата увеличивало пропорцию клеток, гибнущих по апоптотическому пути72. Интересной методической находкой явился подсчет пикселей во флуоресцентных изображениях в зависимости от величины сигнала и спектрального канала регистрации. Это позволило авторам повысить объективность оценки доли живых, погибших от некроза и находящихся в стадии апоптоза нейронов, и тем самым значительно автоматизировать процедуру. Уместно отметить, что оборотной стороной высокой информативности морфологического флуоресцентно-микроскопического метода анализа выживаемости является высокая трудоемкость, главным образом за счет процедуры подсчета клеток. Обнадеживающим обстоятельством можно считать наличие на рынке компьютерных программ для анализа изображений. Недостаток таковых – это, как правило, требование к четкому разделению границ клеток. Это требование относительно легко выполняется в случае таких объектов, как клетки крови, но часто невыполнимо в нейрональных культурах, особенно в культурах высокой плотности и содержащих примесь глии. Указанные программы к тому же довольно дорогостоящи. Ниже приведен сравнительный анализ морфологического подхода и методов, основанных на анализе выживаемости клеточных популяций.
Биохимические методы
Анализ выживаемости клеток по восстановлению солей тетразолия и сопоставление его с методом подсчета клеток, окрашенных флуоресцирующими красителями
Восстановление клетками солей тетразолия до сильно поглощающих в видимой области спектра формазанов послужило основой разработки, пожалуй, самого популярного метода оценки выживаемости клеток. По аббревиатуре, наиболее применяемой для всех солей тетразолия, метод часто называют МТТ-анализом (MTT-assay).
Популярность МТТ-метода обусловлена простотой проведения реакции и легкостью измерения светопоглощения большого количества образцов. Последнее стало возможным благодаря доступности многолуночных плашечных ридеров, представляющих собой фотометры со светофильтрами, набор которых часто определяют сами потребители.
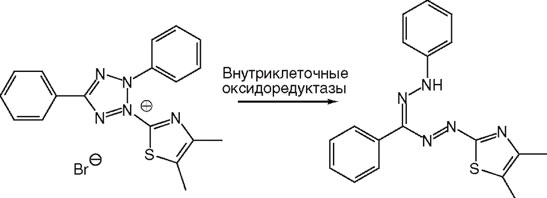
Цвет формазана зависит от структуры соответствующей соли тетразолия и варьирует от синего до красного. Соли тетразолия представляют собой большую группу гетероциклических ароматических соединений, которые часто применяют как для определения выживаемости клеток, так и для установления их окислительновосстановительной способности. Для обозначения солей тетразолия нередко используют аббревиатуры их структурных формул, например МТТ, MTS, NBT, INT, XTT. Формазаны, как правило, плохо растворимы в водных средах и способны образовывать темноокрашенные осадки разного цвета. Известны также соли тетразолия, при восстановлении которых образуются водорастворимые (например, NBT, XTT) и даже флуоресцирующие формазаны (например, СТС73). Структура МТТ и продукта его восстановления до соответствующего формазана представлена на рис. 15 (см. также рис. 23).
Стандартная процедура анализа мало изменилась с того времени, когда она была предложена74 и усовершенствована75, и состоит в инкубации клеток с растворами солей тетразолия, отмывании непрореагировавшего реагента, растворении образовавшегося осадка формазана и фотометрировании окрашенного раствора. При анализе выживаемости нейронов МТТ-методом типичные концентрации МТТ составляют 0,1-0,5 мг/мл, инкубацию проводят обычно при 37 °С. Длительность инкубации зависит от типа нейронов, плотности клеточной культуры, наличия глии и может занимать от 0,5 до 2 часов и более (см., например767778). Растворение кристаллов формазана для последующего фотометрирования предпочтительнее проводить в органических растворителях с относительно низкой летучестью (таких, как ДМСО или изопропанол). Иногда используют смеси детергентов с органическими растворителями (например, 20% додецилсульфата натрия с 40% диметилформамида), в которых клетки и формазан солюбилизируются в течение нескольких часов. Спектр поглощения МТТ-формазана имеет широкую полосу, максимум которой зависит от способа растворения. В ДМСО максимум находится около 530 нм. Для учета поглощения и рассеяния света кюветой спектрофотометра или плашкой, а также растворителем измеряют поглощение в кювете и лунках, заполненных растворителем, либо в тех же лунках, в которых находятся образцы, но при длине волны, при которой формазан уже не поглощает свет (670-700 нм).
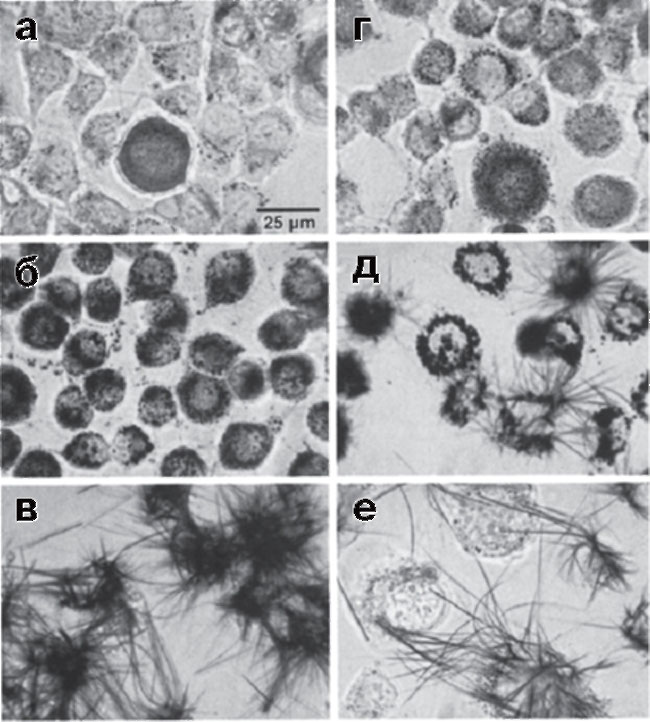
(клеточная линия рака мозга крысы), полученные по мере восстановления МТТ до формазана. Клетки, выращенные в 35-миллиметровых пластиковых чашках, инкубировали с МТТ (0,25 мг/мл, 37 °С) в течение (а) - 5, (б) - 10, (в) - 30, (г) - 90 и (д) - 180 мин, (е) - клетки В12 инкубировали с МТТ 30 мин, затем отмыли невосстановившийся МТТ и продолжили инкубацию в течение еще 180 мин (рисунок воспроизведен из работы Liu et al., 1997)
Несмотря на чрезвычайно широкое применение, механизмы клеточного восстановления солей тетразолия и образования формазанов, даже таких часто используемых, как МТТ, еще не полностью изучены. Во многих статьях (см., например, анализ работ в статьях 798081) и протоколах фирм-производителей внимание акцентируют на восстановлении МТТ в митохондриях и не всегда упоминают другие варианты. Вместе с тем показано, что помимо дегидрогеназ, находящихся в митохондриях8283, соли тетразолия могут восстанавливаться ферментами эндоплазматического ретикулума848586, цитозоля87, короткоживущими внутриклеточными соединениями, например супероксиданион радикалом8889, а также оксидоредуктазами, расположенными на плазматической мембране клеток90. В последнем случае восстановление может происходить во внеклеточном пространстве, если соли тетразолия имеют низкую проницаемость сквозь клеточную мембрану и в среде имеются переносчики восстановительных эквивалентов.
Без ясного понимания механизмов восстановления солей тетразолия и определения внутриклеточных компартментов, где восстановление происходит, трудно понять причину расхождения с данными по выживаемости, получаемыми альтернативными методами, например прямым подсчетом живых и мертвых клеток. Следует отметить, что в большинстве цитированных выше работ идентификацию компартментов проводили на пермибилизованных клетках, их гомогенатах или изолированных субклеточных фракциях. Насколько полученные результаты справедливы для интактных клеток, не всегда ясно. Значительный прогресс в этом направлении был достигнут при применении методов конфокальной и эпифлуоресцентной микроскопии, которые позволили проследить образование формазанов на субклеточном уровне в целых клетках. В работе91, используя изолированные митохондрии из мозга крыс и клетки В12 (опухолевая линия костного мозга), показали, что в изолированных митохондриях малат, глутамат и сукцинат способствуют восстановлению МТТ. Однако в интактных клетках значительная часть формазана образуется в эндосомах и лизосомах (рис. 16, а-г).
Оказалось, что при длительной инкубации эндо- и лизосомальные везикулы с формазаном транспортируются на поверхность клетки, в результате чего во внеклеточном пространстве начинают расти длинные игольчатые кристаллы, превышающие по размерам сами клетки (см. рис. 16, д). О том, что это не внеклеточное восстановление, а именно экзоцитоз, свидетельствует то, что удаление МТТ на стадии, когда образование формазана отмечается только внутри клеток, не отменяло последующего формирования длинных игольчатых кристаллов вне клеток (см. рис. 16, е). Liu и соавт.92 обнаружили, что МТТ не может проникать в клетки В12 за счет диффузии сквозь плазматическую мембрану, а доставляется внутрь клеток в результате эндоцитоза. Эти данные противоречат результатам работы93, в которой на клетках гепатомы HepG2 показано, что хотя не весь МТТ-формазан на конфокальных изображениях колокализуется с митохондриями, часть его оказывается в матриксе, поскольку тушит флуоресценцию потенциалчувствительных митохондриальных зондов. Мы также наблюдали быстрое (в течение 2-3 мин) тушение флуоресценции митохондриального зонда rh123 в культуре гранулярных нейронов мозжечка при очень низких концентрациях МТТ (10 мкг/мл, т.е. около 30 мкМ; Сурин, данные не опубликованы), что свидетельствует о способности МТТ быстро проходить сквозь мембраны, по крайней мере в культивируемых нейронах.

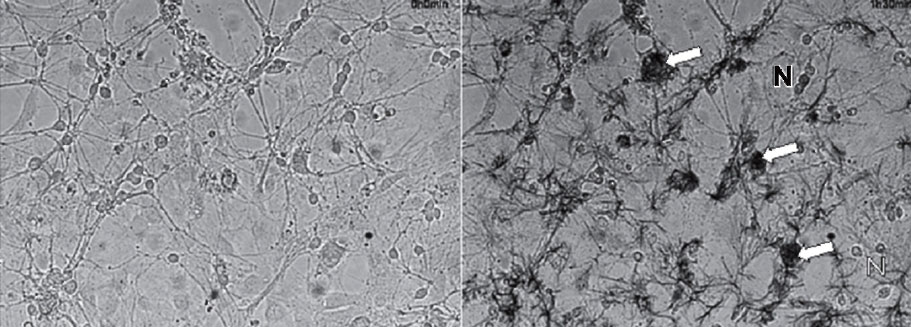
Изображения верхнего ряда показывают, что фаза внутриклеточного образования формазана (среднее изображение) сменяется при длительной инкубации клеток с МТТ-фазой внеклеточного образования игольчатых кристаллов формазана (правое изображение). В нижнем ряду представлены флуоресцентные изображения тех же клеток, показывающие, что во время фазы внеклеточного образования игольчатых кристаллов барьерные свойства клеточной мембраны нейронов резко падают, приводя к накоплению EthD-1. Числа над изображениями соответствуют времени инкубации клеток с МТТ (0,25 мг/мл; комнатная температура). В контрольной культуре, не содержавшей МТТ, окрашивание клеток EthD- 1 за тот же период не изменялось
В культуре нейронов тоже может происходить образование игольчатых кристаллов формазана во внеклеточной среде, если инкубация клеток с МТТ достаточно длительна. На рис. 17 приведены светлопольные и флуоресцентные изображения культуры нейронов мозжечка до МТТ, через полчаса после добавления МТТ, когда темные зерна формазана отмечаются только внутри клеток, и спустя два часа, когда отчетливо видны внеклеточные кристаллы формазана.
Из сопоставления светлопольных изображений с флуоресцентными видно, что фаза интенсивного роста игольчатых кристаллов формазана вне клеток сопровождается резким увеличением внутриклеточной флуоресценции EthD-1. Изображения, полученные при большем увеличении (не представлены), показывают, что нейроны меняют характерную эллиптическую или треугольную форму на шарообразную. Более того, клетки светлеют в светлопольном изображении, теряя образовавшийся внутри них формазан. Эти морфологические изменения наряду с резким увеличением поступления EthD-1 в сому клетки указывают на то, что во время фазы интенсивного роста внеклеточных кристаллов может происходить гибель нейронов. Возможность гибели нейронов под действием образующегося в них формазана, очевидно, необходимо учитывать при интерпретации данных по выживаемости, полученных МТТ-методом.
Нейрональные культуры, даже такие относительно гомогенные, как культуры гранулярных нейронов мозжечка, содержат примесь глиальных клеток, в основном астроцитов.
изображение слева, и через 1,5 ч инкубации при комнатной температуре - изображение справа. Астроциты отмечены светлыми стрелками, нейроны - буквами N
На рис. 18 видно, что в нейрональных культурах, имеющих примесь глиальных клеток (показаны стрелками), значительная доля МТТ может восстанавливаться до формазана не только нейронами (помечены N), но и глией. Вклад глиальных клеток в суммарно образующийся формазан растет, по нашим данным, с увеличением длительности инкубации культуры с МТТ. Причина по крайней мере отчасти связана с большей «живучестью» глиальных клеток. Как отмечалось выше, в фазе роста внеклеточных кристаллов формазана может происходить гибель нейронов (см. рис. 5.17). Гибели глиальных клеток в этом интервале времени не наблюдали. Вероятно, глиальные клетки (астроциты) продолжают восстанавливать МТТ до формазана тогда, когда часть нейронов уже погибла в фазе роста внеклеточных игольчатых кристаллов. Эту особенность смешанных нейрональных культур необходимо учитывать при использовании МТТ-метода для анализа гибели нейронов и, возможно, других типов клеток.
Высвобождение лактатдегидрогеназы как индикатор повреждения и гибели клеток
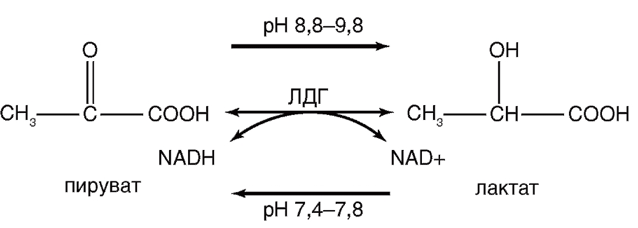
Дегенерация и некроз клеток сопровождаются резким увеличением проницаемости их плазматических мембран для ферментов, являющихся маркерами повреждения. К числу таких ферментов относят дегидрогеназы – оксидоредуктазы, осуществляющие обратимую реакцию окисления/восстановления NAD. Одним из таких ферментов, высвобождающихся во внеклеточную среду в результате нарушения целостности мембраны, является растворимый цитозольный фермент лактатдегидрогеназа (ЛДГ)94, ЛДГ катализирует на последней стадии гликолиза обратимое восстановление пировиноградной кислоты (пирувата) до L-молочной кислоты (лактата) с потреблением в качестве кофермента восстановленной формы NAD – NAD-H (рис. 19).
В уравнении (см. рис. 1) NAD+ и NAD-H – окисленная и восстановленная формы NAD соответственно. ЛДГ окисляет также некоторые другие L-2-гидроксикарбоновые кислоты.
ЛДГ содержится во всех живых организмах, главным образом в цитоплазме клеток. Активной формой ЛДГ является комплекс из четырех субъединиц с молекулярной массой 144 кДа. Каждая субъединица образована пептидной цепью из 334 аминокислот (36 кДа). В организме млекопитающих имеется два типа субъединиц ЛДГ (H и M), незначительно отличающихся по аминокислотной последовательности и способных ассоциировать в тетрамер. Известно 5 изоферментов ЛДГ. Тетрамер состоит либо из одинаковых субъединиц (Н4 или М4), либо из их сочетаний (Н3М, Н2М2, НМ3). В тканях с аэробным обменом (сердце, печень) преобладает тетрамер Н4 (H от англ. heart), характеризующийся максимальной электрофоретической подвижностью среди всех изоферментных форм ЛДГ. В скелетных мышцах преобладает форма М4 (M от англ. muscle), имеющая минимальную электрофоретическую подвижность. Изофермент Н4 (ЛДГ-1) предпочтительно катализирует окисление молочной кислоты в мышечной ткани сердца, а М4 (ЛДГ-5) – восстановление пировиноградной кислоты в скелетных мышцах при низкой концентрации субстрата. ЛДГ-2 (Н3М) содержится по большей части в белых клетках крови; уровень ЛДГ-3 (Н2М2) наиболее высок в легких; ЛДГ-4 (НМ3) преобладает в почках, плаценте и поджелудочной железе.
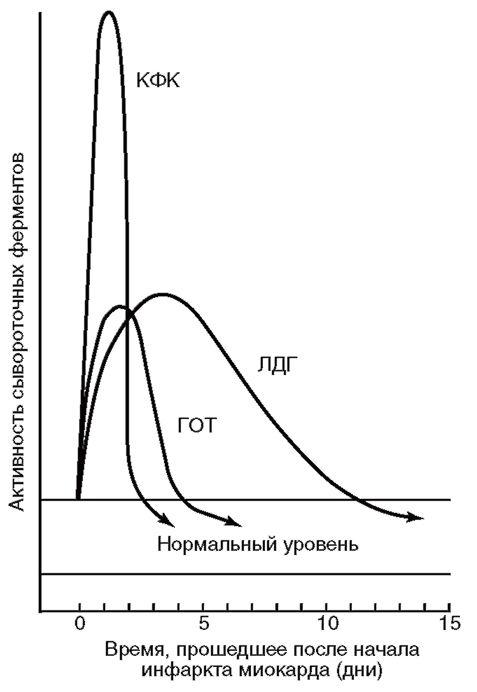
КФК - креатинфосфокиназа; ЛДГ - лактатдегидрогеназа; ГОТ - глутаматоксалоацетаттрансаминаза (цит. по [1])
Все изоформы можно определить в кровотоке. Например, некротизированная во время острого инфаркта миокарда сердечная мышца освобождает в кровь большое количество ферментов. Скорость выброса различных специфических ферментов неодинакова. Изменение уровня ферментов в крови во времени имеет большую диагностическую ценность. Динамика концентрации ферментов, часто используемых для диагностики острого инфаркта миокарда, показана на рис. 20.
В норме в плазме крови уровень ЛДГ-2 выше, чем ЛДГ-1. Повышение уровня ЛДГ-1 по отношению к ЛДГ-2 (flipped pattern» свидетельствует об инфаркте миокарда (повреждение кардиомиоцитов сопровождается высвобождением в кровоток специфической для сердца ЛДГ-1). Уровень ЛДГ-1 нарастает до достижения максимума на 3-4-й день после инфаркта и сохраняется в течение 10 дней, поэтому определение активности ЛДГ в кровотоке используют в медицине как диагностический признак инфаркта.
Для определения повреждения легочной ткани важным диагностическим критерием служит увеличение ЛДГ в бронхоальвеолярном лаваже и в меньшей степени – в плевральной жидкости (Drent et al., 1996). Мониторинг ЛДГ в спинномозговой жидкости также является информативным показателем для ведения больных менингитом95. Кроме того, возрастание уровня ЛДГ наблюдают при гемолитической анемии, гипотензии, в мышцах при повреждении и дистрофии, при ишемии, инфарктах, инфекциях (менингите, энцефалите), гепатите и панкреатите.
Таким образом, повышение активности ЛДГ характерно для широкого спектра патологических состояний, поэтому в клинической практике активность ЛДГ имеет важное диагностическое значение96.
В экспериментах с клеточными культурами или свежеизолированными клетками из органов и тканей измерение ЛДГ используют для оценки повреждений клеток, вызванных действием токсических агентов, а также в экспериментальных моделях инсульта и ишемического состояния. Высвобождение ЛДГ во внеклеточную среду возможно при морфологических и функциональных повреждениях клеток97. Таким образом, уровень активности ЛДГ в среде культивируемых клеток служит показателем цитотоксичности.
Принцип метода
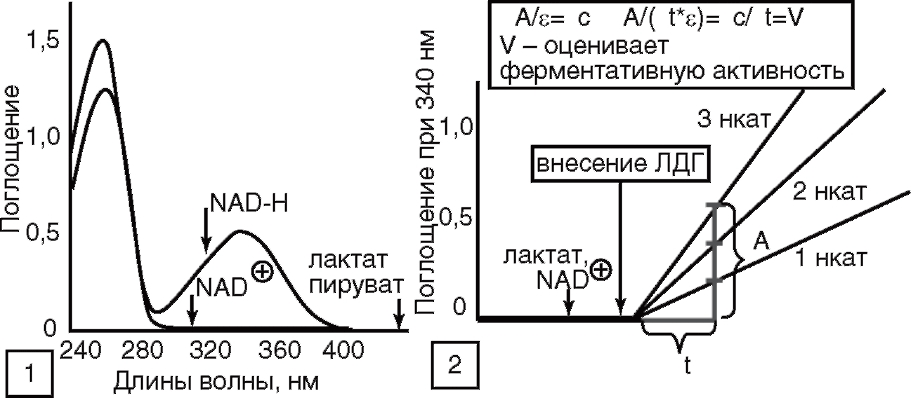
(Кольман, Рём, 2000)
Анализ активности ЛДГ как в биологических жидкостях организма в клинической практике, так и в среде культивирования клеток в экспериментальных условиях базируется на способности фермента катализировать окисление лактата до пирувата с восстановлением NAD+ до NAD-H. Восстановленный кофермент NAD-H поглощает свет при 340 нм, в то время как у NAD+ при этой длине волны поглощение отсутствует (рис. 21, 1). Различия в поглощении NAD+ и NAD-H между 300 и 400 нм обусловлены длинноволновым сдвигом полосы поглощения никотинамидного кольца при его восстановлении. Для определения активности в кювету помещают растворы лактата и NAD+, добавляют фермент и регистрируют увеличение поглощения ДА при длине волны 340 нм (рис. 21, 2), соответствующей максимуму в УФ-спектре NAD-H (см. рис. 21, 1). Некаталитическая реакция протекает с очень низкой скоростью, и измеряемые количества NAD-H образуются только после добавления ЛДГ. Так как скорость увеличения поглощения ΔА/Δt по закону Ламберта-Бера пропорциональна скорости реакции Δc/Δt (см. рис. 21, 2), активность ЛДГ можно рассчитать с помощью молярного коэффициента экстинкции ε при 340 нм или путем сравнения со стандартным раствором. Данный метод определения активности ЛДГ (с использованием NAD-H в качестве маркера реакции) чаще применяют в клинической практике для анализа небольшого количества образцов достаточного объема. На этом принципе основана работа биохимических наборов для определения активности ЛДГ, предлагаемых фирмами RANDOX, UK (www.randox.com) и Diagnostic Chemicals Ltd. (Канада). Производители указывают активность ЛДГ в разных биологических жидкостях человека в норме. В клинических условиях используют абсолютные значения активности ЛДГ, например в сыворотке крови, оценивая данный показатель в динамике и сопоставляя его со значениями активности фермента в норме.
Описание метода
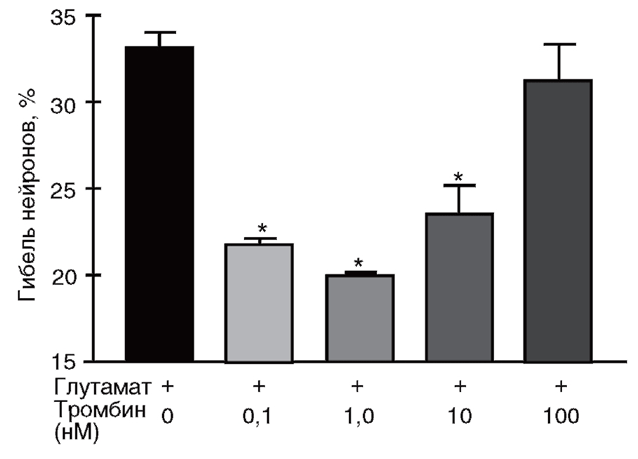
вызванную 15-минутной инкубацией культур со 100 мкМ глутамата. Гибель оценивали по высвобождению ЛДГ из клеток в среду через 24 ч после действия глутамата. *р <0,05 по сравнению с глутаматом (Горбачева и др., 2006)
Для оценки выживаемости клеток необходимо определить не только активность фермента, высвободившегося во внеклеточный буфер, но и общую активность ЛДГ в культуре клеток (в контроле). Для этого сравнивают активность фермента во внеклеточной среде с активностью ЛДГ в клеточном лизате. Например, при оценке протекторного действия низких концентраций тромбина в условиях токсического воздействия глутамата на нейроны (рис. 22) клеточные культуры инкубировали в присутствии тромбина и 100 мкМ глутамата. Для определения процента гибели нейронов через 24 ч после воздействия образцы культуральной среды отбирали для анализа активности ЛДГ (50 мкл=5% от объема среды в лунке или чашке). Затем клетки лизировали 0,2% раствором детергента «Тритон Х-100» в течение 15 мин при 37 °С (либо 2-3-кратным замораживанием/оттаиванием). Лизат центрифугировали, супернатант отбирали для определения в нем активности ЛДГ. Процент гибели нейронов рассчитывали как (Активность ЛДГ в клеточной среде/Общая величина ЛДГ) х 100, (5.4) где общая величина ЛДГ определялась после лизиса клеток.
Обычно используемые для определения активности ЛДГ тесты предполагают плотность исследуемых клеточных культур на уровне 104-106 клеток на ячейку. Однако появление биохимических наборов для определения ЛДГ, основанных на двухэтапном процессе восстановления, позволило повысить чувствительность метода. В этом варианте NAD-H, образовавшийся при восстановлении NAD+ путем окисления лактата в пируват, снова окисляется, но уже с образованием либо сильно поглощающих в видимой области спектра, либо флуоресцирующих веществ. Содержание этих конечных маркеров определяют колориметрически или флуориметрически. В качестве конечного маркера реакции, катализируемой ЛДГ, может выступать формазан, образованный из тетразолиума при участии NAD-H (рис. 23) или флуоресцирующее вещество (рис. 24).
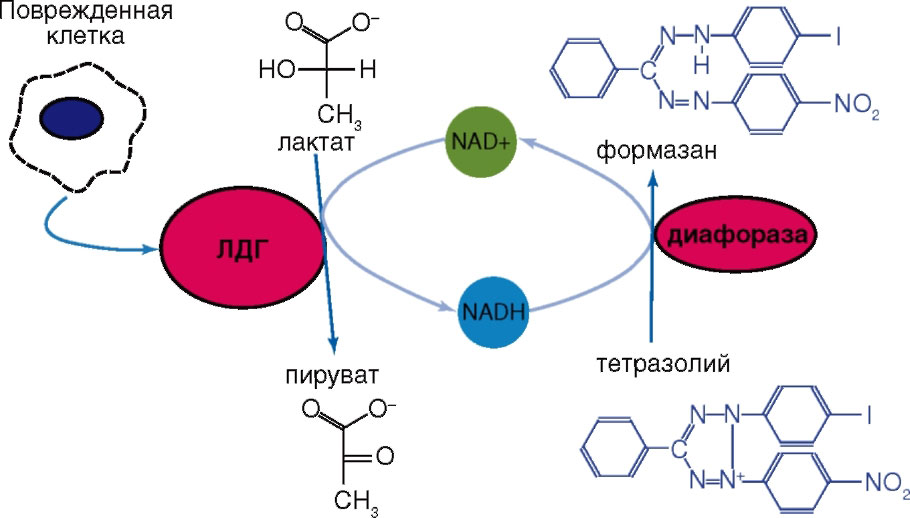
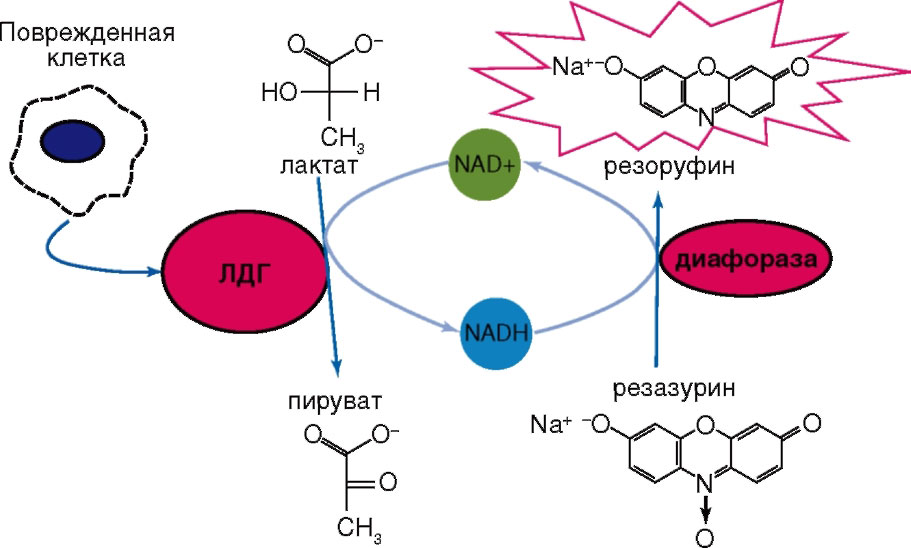
Реакция образования формазана лежит в основе методов, предлагаемых для определения активности ЛДГ, например, фирмами G-Bioscience, Oxford Biomedical Research (www.oxfordbiomed.com), Cayman Chemical Company (www.caymanchem.com), Roche Diagnostics Corporation (www.roche.com). SensoLyte Cell Cytotoxicity Assay Kit (www. anaspec.com); в ней используют резоруфин (590 нм) как флуоресцентный индикатор для измерения активности ЛДГ (см. рис. 24).
Недостатки и преимущества ЛДГ-метода
Недостатком ЛДГ-метода является ограничение использования культур клеток, среда которых включает бычью сыворотку (Fetal Bovine Serum, FBS). FBS содержит ЛДГ, что может искажать результаты эксперимента, поэтому в данном случае рекомендуют применять культуральные среды с низким уровнем FBS (менее 1%) или среды, свободные от него. В случаях, когда культуральная среда содержит высокий процент FBS (более 10%), необходимо измерять контрольные образцы, содержащие культуральную среду с FBS.
Метод определения гибели клеток по уровню высвобождения ЛДГ имеет ряд преимуществ по сравнению с другим биохимическим методом оценки гибели клеток – МТТ-тестом:
- во-первых, ЛДГ-метод обладает высокой точностью – получаемые с его помощью показатели гибели клеток меньше зависят от различий в плотности клеток в культурах, поскольку оценивают как активность поврежденных клеток, так и общую активность каждой отдельной культуры. В МТТ-тесте показатели гибели клеток в обработанных культурах (абсорбционность растворов формазана) всегда сопоставляются с контрольной культурой, не подвергавшейся воздействию;
- во-вторых, ЛДГ-метод позволяет исследовать временную динамику клеточной гибели в условиях одной культуры путем отбора проб среды, в которой культивируются исследуемые клетки в разные временные интервалы. С помощью МТТ-теста данную манипуляцию можно осуществить, только отбирая (изымая) для анализа отдельные культуры клеток в определяемые интервалы времени.
Footnotes
- Berridge M.J., Lipp P., Bootman M.D. The versatility and iniversality of calcium signaling // Nature Rev. – 2000. – Vol. 1. – P. 11-21.
- Berridge M.J., Bootman M.D, Roderick H.L. Calcium signaling: Dynamics, homeostasis and remodellinf // Nature Rev. – 2003. – Vol. 4. -517-529.
- Chiesa A., Rapizzi E, Tosello V. et al. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signaling //Biochem. J. – 2001. – Vol. 355. – P. 1-12.
- Devos D., Dokudovskaya S., Williams R. et al. Simple fold composition and modular architecture of the nuclear pore complex // Proc. Acad.Sci. (USA). – 2006. – Vol. 103. – P. 2172-2177.
- Porcelli A.M., Ghelli A., Zanna C. et al. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant // Biochem. Res. Commun. – 2005. – Vol. 326. – P. 799-804.
- Ridgeway E.B., Ashley C.C. Calcium transients in single muscle fibers // Biophys. Res. Commun. – 1967. – Vol. 29. – P. 229-234.
- Rizzuto R., Pozzan T. Microdomains of intracellular Са2+: molecular determinants and functional consequences // Physiol. – 2006. -Vol. 86. – P. 369-408.
- Rizzuto R., Pinton P., Carrington W. et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Са2+ responses // Science. – 1998. – Vol. 280. – P. 1763 – 1766.
- Heo D.S., Park J.G., Hata K. et al. Evaluation of tetrazolium-based semiautomatic colorimetric assay for measurement of human antitumor cytotoxicity // Cancer Res. – 1990. – Vol. 12. – P. 3681-3690.
- O’Malley D.M. Calcium permeability of the neuronal nuclear envelope: evaluation using confocal volumes and intracellular perfusion // J. Neurosci. – 1994. – Vol. 14. – P. 5741-5758.
- Veloso D., Guynn R.W., Oskarson M., Veech R.L. The concentrations of free and bound magnesium in rat tissues. Relative constancy of free Mg2+ concentrations // J. Biol. Chem. – 1973. – Vol. 248. – P. 4811-4819.
- Chiesa A., Rapizzi E, Tosello V. et al. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signaling //Biochem. J. – 2001. – Vol. 355. – P. 1-12.
- Takahashi S., Abe T., Gotoh J., Fukuuchi Y. Substrate-dependence of reduction of MTT: a tetrazolium dye differs in cultured astroglia andneurons // Neurochem. Int. – 2002. – Vol. 40. – P. 441-448.
- Николс Д.Г., Мартин А.Р., Валлас Б.Д., Фукс П.А. От нейрона к мозгу. 4-е изд. – Изд. научной и учебной литературы УРСС, 2003. – 671 с.
- Nicholls D.G. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease // Int. J. Biochem. Cell Biol. – 2002. – Vol. 34. – P. 1372-1381.
- Nicholls D.G. Mitochondria and calcium signaling // Cell Calcium. – – Vol. 38. – P. 311-317.
- Gerencser A.A., Adam-Vizi V. Selective, high-resolution fluorescence imaging of mitochondrial Са2+ concentration // Cell Calcium. – 2001. -Vol. 30. – P. 311-321.
- Krieger C., Duchen M.R. Mitochondria, Са2+ and neurodegenerative disease // Eur. J. – 2002. – Vol. 447. – P. 177-188.
- Kroemer G., El-Deiry W.S., Golstein P. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death // Cell Death Differ. – 2005. – Vol. 12. – P. 1463-1467.
- Nicholls D.G. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease // Int. J. Biochem. Cell Biol. – 2002. – Vol. 34. – P. 1372-1381.
- Nicholls D.G., Chalmers S. The integration of mitochondrial calcium transport and storage // J. Bioenerg. – 2004. – Vol. 36. – P. 277-281.
- O’Malley D.M. Calcium permeability of the neuronal nuclear envelope: evaluation using confocal volumes and intracellular perfusion // J. Neurosci. – 1994. – Vol. 14. – P. 5741-5758.
- Rink T.J., Tsien R.J., Pozzan T.J. Cytoplasmic pH and free Mg2+ in lymphocytes // Cell Biol. – 1982. – Vol. 95. – P. 189-196.
- Tsien R.Y. Fluorescent indicators of ion concentrations // Methods Cell Biol. – 1989. – Vol.- P. 127-156.
- Tarpey M.M., Fridovich I. Methods of Detection of Vascular Reactive Species Nitric Oxide, Superoxide, Hydrogen Peroxide, and Peroxynitrite // Circ. Res. – 2001. – Vol. 89. – P. 224-236.
- Toescu E.C., Verkhratsky A., Landfield P.W. Ca2+ regulation and gene expression in normal brain aging // Tends in Neurosci. – 2004. -Vol. 27. – P 614-620.
- Duchen M.R., Verkhratsky A., Muallem S. Mitochondria and calcium in health and disease // Cell Calcium. – 2008. – Vol. 44.- P. 1-5.
- Golovina V.A., Blaustein M.P. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum // Science. – 1997. -Vol. 275. – P. 1643-1648.
- Goodwin C.J., Holt S.J., Riley P.A. et al. Growth hormone-responsive DT-diaphorase-mediated bioreduction of tetrazolium salts // Biochem. Biophys. Res. Commun. – 1996. – Vol.226- P. 935-941.
- Gusev E.I., Skvortsova V.I. Glutamate neurotransmission and calcium metabolism in cerebral ischaemia and under normal conditions // Usp. Nauk. (Rus). – 2002. – Vol. 33. – P. 80-93.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays // J. Immunol. – 1983. – Vol. 65. – P. 55-63.
- Toescu E.C., Verkhratsky A., Landfield P.W. Ca2+ regulation and gene expression in normal brain aging // Tends in Neurosci. – 2004. -Vol. 27. – P 614-620.
- Tsien R.Y. Fluorescent probes of cell signaling // Annu. Neurosci. – 1989. – Vol. 12. – P. 227-253.
- Veloso D., Guynn R.W., Oskarson M., Veech R.L. The concentrations of free and bound magnesium in rat tissues. Relative constancy of free Mg2+ concentrations // J. Biol. Chem. – 1973. – Vol. 248. – P. 4811-4819.
- Boitier E, Rea R., Duchen M.R. Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes //Cell Biol. – 1999. – Vol. 145. – P. 795-808.
- Devos D., Dokudovskaya S., Williams R. et al. Simple fold composition and modular architecture of the nuclear pore complex // Proc. Acad.Sci. (USA). – 2006. – Vol. 103. – P. 2172-2177.
- Rizzuto R., Pinton P., Carrington W. et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses // Science. – 1998. – Vol. 280. – P. 1763 – 1766.
- Davidson S.M., Duchen M.R. Calcium microdomains and oxidative stress // Cell Calcium. – 2006. – Vol. 40. – P. 561-574.
- Golovina V.A., Blaustein M.P. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum // Science. – 1997. -Vol. 275. – P. 1643-1648.
- Baird G.S., Zacharias D.A., Tsien R.Y. Circular permutation and receptor insertion within green fluorescent proteins // Proc. Acad. Sci. (USA). – 1999. – Vol. 96. – P. 11241-11246.
- Belousov V.V., Fradkov A.F., Lukyanov K.A. et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide // Nat. Methods. — Vol. 3. – P. 281-286.
- Manev H., Kharlamov E., Uz T. et al. Characterization of zinc-induced neuronal death in primary cultures of rat cerebellar granule cells // Exp. Neurol. – 1997. – Vol. 146. – P. 171-178.
- Gerencser A.A., Adam-Vizi V. Selective, high-resolution fluorescence imaging of mitochondrial Ca2+ concentration // Cell Calcium. – 2001. -Vol. 30. – P. 311-321.
- Rizzuto R., Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences // Physiol. – 2006. -Vol. 86. – P. 369-408.
- Scheuer J., Berry M.N. Effect of alkalosis on glycolysis in the isolated rat heart // Am. J. – 1967. – Vol. 213. – P. 1143-1148.
- Gerencser A.A., Adam-Vizi V. Selective, high-resolution fluorescence imaging of mitochondrial Ca2+ concentration // Cell Calcium. – 2001. -Vol. 30. – P. 311-321.
- Diaz G., Melis M., Musin A. et al. Localization of MTT formazan in lipid droplets. An alternative hypothesis about the nature of formazan granules and aggregates // Eur. J. Histochem. – 2007. – Vol. 51. – P. 213-218.
- Leist M., Volbracht C., Kiihnle S. et al. Caspase-mediated apoptosis in neuronal excitotoxicity triggered by nitric oxide // Mol. Med. – 1997. -Vol. 3. – P. 750-764.
- Orrenius S., Zhivotovsky B., Nicotera P. Regulation of cell death: The calcium-apoptosis link // Nature Rev. – 2003. – Vol. 4. – P. 552-565.
- Petyaev I.M., Hunt J.V. Micellar acceleration of oxygen-dependent reactions and its potential use in the study of human low density lipoprotein // Biochim. Acta. – 1997. – Vol. 1345. – P. 293-305.
- Bolshakov A.P., Mikhailova M.M., Szabadkai G. et al. Measurements of mitochondrial pH in cultured cortical neurons clarify contribution of mitochondrial pore to the mechanism of glutamate-induced delayed Ca2+ deregulation // Cell Calcium. – 2008 .- Vol. 43. – P. 602-614.
- Gusev E.I., Skvortsova V.I. Glutamate neurotransmission and calcium metabolism in cerebral ischaemia and under normal conditions // Usp. Nauk. (Rus). – 2002. – Vol. 33. – P. 80-93.
- Krieger C., Duchen M.R. Mitochondria, Ca2+ and neurodegenerative disease // Eur. J. – 2002. – Vol. 447. – P. 177-188.
- Nicholls D.G. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease // Int. J. Biochem. Cell Biol. – 2002. – Vol. 34. – P. 1372-1381.
- Krieger C., Duchen M.R. Mitochondria, Ca2+ and neurodegenerative disease // Eur. J. – 2002. – Vol. 447. – P. 177-188.
- Nicholls D.G. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease // Int. J. Biochem. Cell Biol. – 2002. – Vol. 34. – P. 1372-1381.
- Szabadkai G., Duchen M.R. Mitochondria: the hub of cellular Ca2+ signaling // Physiology (Bethesda). – 2008. – Vol. 23. – P. 84-94.
- Wang X., Mori T., Sumii T., Lo E.H. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress //Stroke. – 2002. – Vol. 33. – P. 1882-1888.
- Rizzuto R., Bernardi P., Pozzan T. Mitochondria as all-round players of the calcium game // J. Physiol. – 2000. – Vol. 529. – P. 37-47.
- Alonso M.T., Villalobos C, Chamero P. et al. Calcium microdomains in mitochondria and nucleus // Cell Calcium. – 2006. – Vol. 40. – P. 513-525.
- Butt A.A., Michaels S., Greer D. et al. Serum LDH level as a clue to the diagnosis of histoplasmosis // AIDS Read. – 2002. – Vol. 12. – P. 317-321.
- Pereira C., Santos M.S., Oliveira C. Metabolic inhibition increases glutamate susceptibility on a PC12 cell line // J. Neurosci. – 1998. – Vol. 51. – P. 360-370.
- Bolshakov A.P., Mikhailova M.M., Szabadkai G. et al. Measurements of mitochondrial pH in cultured cortical neurons clarify contribution of mitochondrial pore to the mechanism of glutamate-induced delayed Ca2+ deregulation // Cell Calcium. – 2008 .- Vol. 43. – P. 602-614.
- Liu Y., Peterson D.A., Kimura H., Schubert D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction // J. Neurochem. – 1997. – Vol. 69. – P. 581-593.
- Liu Y., Peterson D.A., Kimura H., Schubert D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction // J. Neurochem. – 1997. – Vol. 69. – P. 581-593.
- Liu Y., Peterson D.A., Kimura H., Schubert D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction // J. Neurochem. – 1997. – Vol. 69. – P. 581-593.
- Ankarcrona M, Dypbukt J.M., Bonfoco E. et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function // Neuron. – 1995. – Vol. 15. – P. 961-973.
- Kamat D.V., Chakravorty B.P. Comparative values of CSF-LDH isoenzymes in neurological disorders // Ind. J. Med. Sci. – 1999. – Vol. 53. -1-6.
- Baskic D, Popovic S, Ristic P., Arsenijevic N.N. Analysis of cycloheximideinduced apoptosis in human leukocytes: fluorescence microscopy using annexin V/propidium iodide versus acridin orange/ethidium bromide // Cell Biol. Int. – 2006. – Vol. 30. – P. 924-932.
- Liu Y., Peterson D.A., Kimura H., Schubert D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction // J. Neurochem. – 1997. – Vol. 69. – P. 581-593.
- Baskic D, Popovic S, Ristic P., Arsenijevic N.N. Analysis of cycloheximideinduced apoptosis in human leukocytes: fluorescence microscopy using annexin V/propidium iodide versus acridin orange/ethidium bromide // Cell Biol. Int. – 2006. – Vol. 30. – P. 924-932.
- Minta A., Kao J.P., Tsien R.Y. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores // J. Biol. – 1989. – Vol. 264. – P. 8171-8178.
- Bernas T., Dobrucki J.W. The role of plasma membrane in bioreduction of two tetrazolium salts, MTT, and CTC // Arch. Biophys. – 2000. – Vol. 380. – P. 108-116.
- Nicholls D.G., Budd S.L. Mitochondria and neuronal survival // Physiol.Rev. – 2000. – Vol. 80. – P. 315-360.
- Denizot F., Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability // J. Immunol. – 1986. – Vol. 89. -271-277.
- Marshall N.J., Goodwin C.J., Holt S.J. A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function //Growth Regul. – 1995. – Vol. 5. – P. 69-84.
- Mironova E.V., Evstratova A.A., Antonov S.M. A fluorescence vital assay for the recognition and quantification of excitotoxic cell death by necrosis and apoptosis using confocal microscopy on neurons in culture // J. Neurosci. – 2007. – Vol. 163. – P. 1-8.
- Takahashi A., Camacho P., Lechleiter J.D., Herman B. Measurement of intracellular calcium // Physiol. – 1999. – Vol. 79. – P. 1089-1125.
- Bernas T., Dobrucki J.W. The role of plasma membrane in bioreduction of two tetrazolium salts, MTT, and CTC // Arch. Biophys. – 2000. – Vol. 380. – P. 108-116.
- Bernas T., Dobrucki J. Mitochondrial and nonmitochondrial reduction of MTT: interaction of MTT with TMRE, JC-1, and NAO. Mitochondrial fluorescent probes // Cytometry. – 2002. – Vol. 47. – P. 236-242.
- Lee M.A., Dunn R.C., Clapham D.E., Stehno-Bittel L. Calcium regulation of nuclear pore permeability // Cell Calcium. – 1998. – Vol. 23. -91-101.
- Berridge M.V., Tan A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT):subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction // Arch. Biochem. Biophys – 1993. – Vol. 303. – P. 474-482.
- Izumi Y., Izumi M, Benz A.M., Zorumski Ch.F. Lactate dehydrogenase release is facilitated by brief sonication of rat hippocampal slices and isolated retinas following acute neuronal damage // J. Neurosci. – 2001. – Vol. 108. – P. 49-55.
- Berridge M.V., Tan A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT):subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction // Arch. Biochem. Biophys – 1993. – Vol. 303. – P. 474-482.
- Grynkiewicz G, Poenie M, Tsien R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties // J. Biol. – 1985. – Vol. 260. – P. 3440-3450.
- Perez-Terzic C., Stehno-Bittel L, Clapham D.E. Nucleoplasmic and cytoplasmic differences in the fluorescence properties of the calcium indicator Fluo-3 // Cell Calcium. – 1997. – Vol.21- P. 275-282.
- Berridge M.V., Tan A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT):subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction // Arch. Biochem. Biophys – 1993. – Vol. 303. – P. 474-482.
- Reynolds I.J., Malaiyandi L.M., Coash M., Rintoul G.L. Mitochondrial trafficking in neurons: a key variable in neurodegeneration? // J. Bioenerg.Biomembr. – 2004. – Vol. 36. – P. 283-286.
- Verkhratsky A. Physiology and Pathophysiology of the Calcium Store in the Endoplasmic Reticulum of Neurons // Physiol. – 2005. -Vol. 85. – P. 201-279.
- Bernas T., Dobrucki J. Mitochondrial and nonmitochondrial reduction of MTT: interaction of MTT with TMRE, JC-1, and NAO. Mitochondrial fluorescent probes // Cytometry. – 2002. – Vol. 47. – P. 236-242.
- Lee M.A., Dunn R.C., Clapham D.E., Stehno-Bittel L. Calcium regulation of nuclear pore permeability // Cell Calcium. – 1998. – Vol. 23. -91-101.
- Liu Y., Peterson D.A., Kimura H., Schubert D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction // J. Neurochem. – 1997. – Vol. 69. – P. 581-593.
- Bernas T., Dobrucki J. Mitochondrial and nonmitochondrial reduction of MTT: interaction of MTT with TMRE, JC-1, and NAO. Mitochondrial fluorescent probes // Cytometry. – 2002. – Vol. 47. – P. 236-242.
- Bonfoco E, Krainc D, Ankarcrona M. et al. Apoptosis and necrosis: Two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures // Proc. Acad. Sci. (USA). – 1995. – Vol. 92. – P. 7162-7166.
- Khodorov B. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurons // Prog.Biophys. Mol. Biol. – 2004. – Vol. 86. – P. 279-351.
- Carafoli E. Calcium signaling: A tale for all seasons // Proc. Acad.Sci. (USA). – 2002. – Vol. 99. – P. 1115-1122.
- Jekabsons M.B., Nicholls D.G. Bioenergetic analysis of cerebellar granule neurons undergoing apoptosis by potassium/serum deprivation // Cell Death Differ. – 2006. – Vol. 13. – P. 1595-1610.